Enterobase nomenclature
nomenclature is a tool nested within The Calculation Engine platform and can handle all the requests from EnteroBase website or from EnteroBase Backend Pipeline pipelines. It currently stores all the MLST information by itself (although NServ provides an alternative way to query the underlying database). It can be called independently and is generally run (for each available genotyping scheme) after the :doc:’QAssembly <backend-pipeline-qassembly>’ pipeline, which offers an automatic solution from short reads to MLST typing. nomenclature currently is in version 3.2.
Identifying allele coordinates
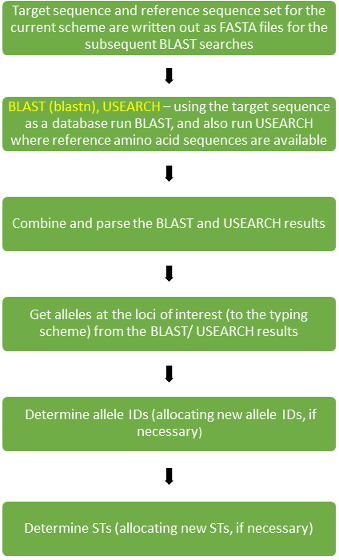
For a given genotyping scheme, there is a reference set (“refset”) of sequences that are used to identify the allele coordinates in a target sequence relevant to the loci in the scheme. (The target sequence may either be from genome sequence assembled from uploaded reads or a sequence of interest uploaded for a Locus Search. In the latter case nomenclature will have been invoked via the locus_search pipeline.) In the case of loci with (protein) coding sequence then both the nucleotide and amino acid sequence will be used. (Some genotyping schemes may include some loci that code for special cases of RNA sequence, such as ribosomal RNA, or that are non-coding.) A BLAST (version 2.2.31) search is carried out (using blastn) with the nucleotide reference sequences and a USEARCH (version 8.0.1623) search is carried out with any available amino acid references sequences. The search results are combined and parsed and from this the alleles at the loci of interest in the target sequences are identified.
Assigning allele IDs
Allele sequences are looked up in the database in order to determine the allele ID. If the allele is fragmented or duplicated it is assigned a negative value (with the specific value being essentially meaningless) and if it is missing then it assigned a zero. Given a novel allele sequence, one of two things can happen. If the allele sequence is of sufficient quality and trusted then the new allele sequence may be added to the database and assigned an allele ID. The new allele ID will be assigned the value of the current largest value allele ID for that locus incremented by 1. An otherwise potential new allele sequence - if not trusted, as is typical in Locus Search, for example - may be treated either as a fragmented/ duplicated or missing locus case and will not result in a new allele sequence being added to the database.
Assigning STs
The set of allele IDs (with the alleles corresponding to loci in the genotyping scheme that is used) found for a genome assembly (or uploaded sequence from :doc:`Locus Search </features/locus-search>`_) is looked up in the database in order to determine the ST. Given a novel set of allele IDs, a similar situation applies to that for a novel allele ID and one of two things can happen. If genome assembly/ sequence giving the novel set of allele IDs is of sufficient quality and trusted then the new set of allele IDs is added to the database and assigned an ST. The new ST will be assigned the value of the current largest ST value incremented by 1. Otherwise the ST is flagged as problematic and indeterminate.
Inputs/Outputs
Parameters
{
"params": {
"scheme": "Senteria_UoW", # Ecoli_UoW, Yersinia_UoW, Mcatarrhalis_UoW
"genome_id": "SAL_AA0001AA_AS"
},
"inputs": {
"genome_seq": "SAL_AA0001AA_AS.scaffold.fastq"
}
}
Output fields
History
Version 3.2
Support added for new allele and ST callings from other MLST schemes.
Version 2.0
Starts to call new ST types from MLST UoW.
Version 1.0
Serves new allele callings from MLST UoW.