Backend pipelines
The EnteroBase pipeline suite currently functions via four “black boxes”: an engine for downloading and parsing metadata (MetaParser), a bulk downloading engine (RCatch), a calculation engine (The Calculation Engine) and a separate nomenclature server (NServ) which are accessible via APIs that communicate with the EnteroBase website. The website will also offer an API interface for access by external computers, but that is not yet available. The EnteroBase pipelines and four “black boxes” are written in Python 3.8 with a PostgreSQL 10/13 database for storing information. Source code is available upon request.
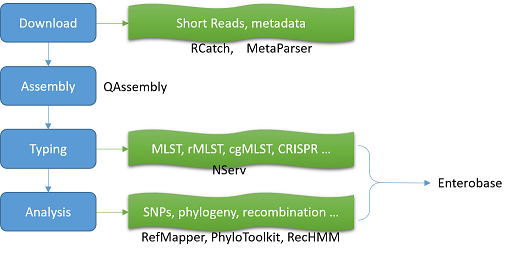
About the components
About the databases
Pipelines
Workflow after receiving reads
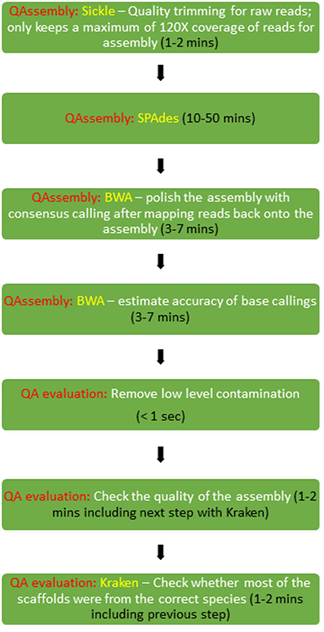
1. Automatic assembly
QAssembly - consists of sequential processing in 3 main steps consisting of the assembly itself in [QAssembly], [QAtoFasta] and [QA evaluation]. - Assembly in QAssembly - a one-stop solution from short reads to high
quality assemblies, including read pre-processing, trimming, assembly, post-correction and filtering.
QAtoFasta - conversion of assemblies from QAFastq format to Fasta format
QA evaluation - evaluates the quality of assemblies based on multiple criteria. (Assemblies that fail this quality control will not be used to call MLST or in other downstream analyses).
2. MLST typing - nomenclature - after an assembly has been carried out and passed the QC criteria MLST typing
will be done using all of the available schemes for the relevant genus/ database.
3. Annotation - prokka_annotation - also after successful assembly, the [prokka_annotation] pipeline is run.
4. Serovar Prediction - SeroPred - the SeroPred pipeline is run after successful assembly in the case of the Salmonella
and Escherichia/Shigella databases, in order to predict the serovar from the assembled sequences (using SISTR in the case of the Salmonella database).
Minimum Spanning Trees
DMSTree - the minimum spanning tree pipeline is called DMSTree, for computing a minimum spanning tree
for strains based on a chosen genotyping scheme. It will be fully described in a research paper that is currently in preparation.
SNP Trees
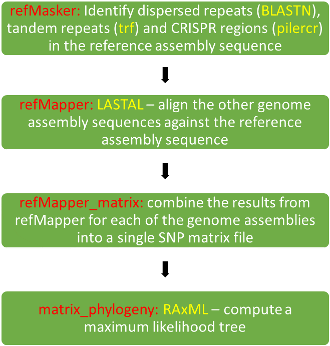
SNP Tree pipelines - a combined pipeline/ workflow in EnteroBase runs a number of pipeline jobs with The Calculation Engine (TCE) in the order refMasker, refMapper, refMapper_matrix, matrix_phylogeny. Earlier steps in the workflow are skipped if the pipeline has been previously run on the same data - refMasker is not run if it has already been run on the given reference assembly, assemblies that already have SNPs called against the chosen reference are not re-processed in refMapper (potentially meaning that refMapper is not run).
Locus Search
locus_search - when the locus_search pipeline is started - by use of Locus Search on the EnteroBase website, TCE actually runs the nomenclature pipeline (for the scheme chosen in the Locus Search). However, unlike an MLST typing job with nomenclature on an uploaded assembly, no new allele IDs or STs will be assigned in the event of a locus being discovered with a novel allele, since the quality of sequence uploaded via the Locus Search page cannot be ascertained.
Other EnteroBase Internal Components
MetaParser
MetaParser implements - the automated downloading of all GenInfo Identifiers (GI numbers) in
NCBI Short Read Archives (SRAs) or complete or partial assemblies with the genus designation Salmonella, Escherichia / Shigella, Yersinia or Moraxella, and the corresponding metadata (via ENTREZ utilities)
parsing of the metadata into a consistent, EnteroBase format
RCatch Given a GI number RCatch downloads the corresponding short read archive from any of three major public sequence databases
(SRA/NCBI [Short Read Archives (SRAs)], ENA/EBI and DRA/DDBJ)
The Calculation Engine
The Calculation Engine (TCE) is a system that runs a number of pipelines including those that assemble short read archives or user uploaded reads, evaluate and modify the assemblies, and pass the final assemblies onto [nomenclature] for genotyping and also computation of minimum spanning trees based on the genotyping schemes and SNP trees. The system has a queue for the different jobs running the pipelines, support for job management and support for distributing the different jobs between multiple computer systems.
NServ
NServ genotypes genomes from assembly data. It currently handles the 7-gene MLST, rMLST, cgMLST and wgMLST (and also the 8 gene classic MLST scheme used by Moraxella. NServ does automatic nomenclature for all new genomes coming in, and concurrently synchronises 7-gene MLST with the main MLST web site and rMLST with the rMLST web site.