EnteroBase Internal Structure (Website)
Persistent data in EnteroBase is split across several PostGresSQL databases. The website databases store User information and strain-centric information. NServ stores information pertaining to typing schemes, such as information about alleles, sequences and STs. For more information about NServ, see EnteroBase Database Structure (NServ).
System Database Structure
The system database (Figure 1) stored User information, their preferences, and their uploaded reads (before it is processed). Thier sharing settings with their ‘buddies’ are stored across UserBuddies (which defines who is a buddy) and BuddyPermissionTags (which defines buddy permissions).
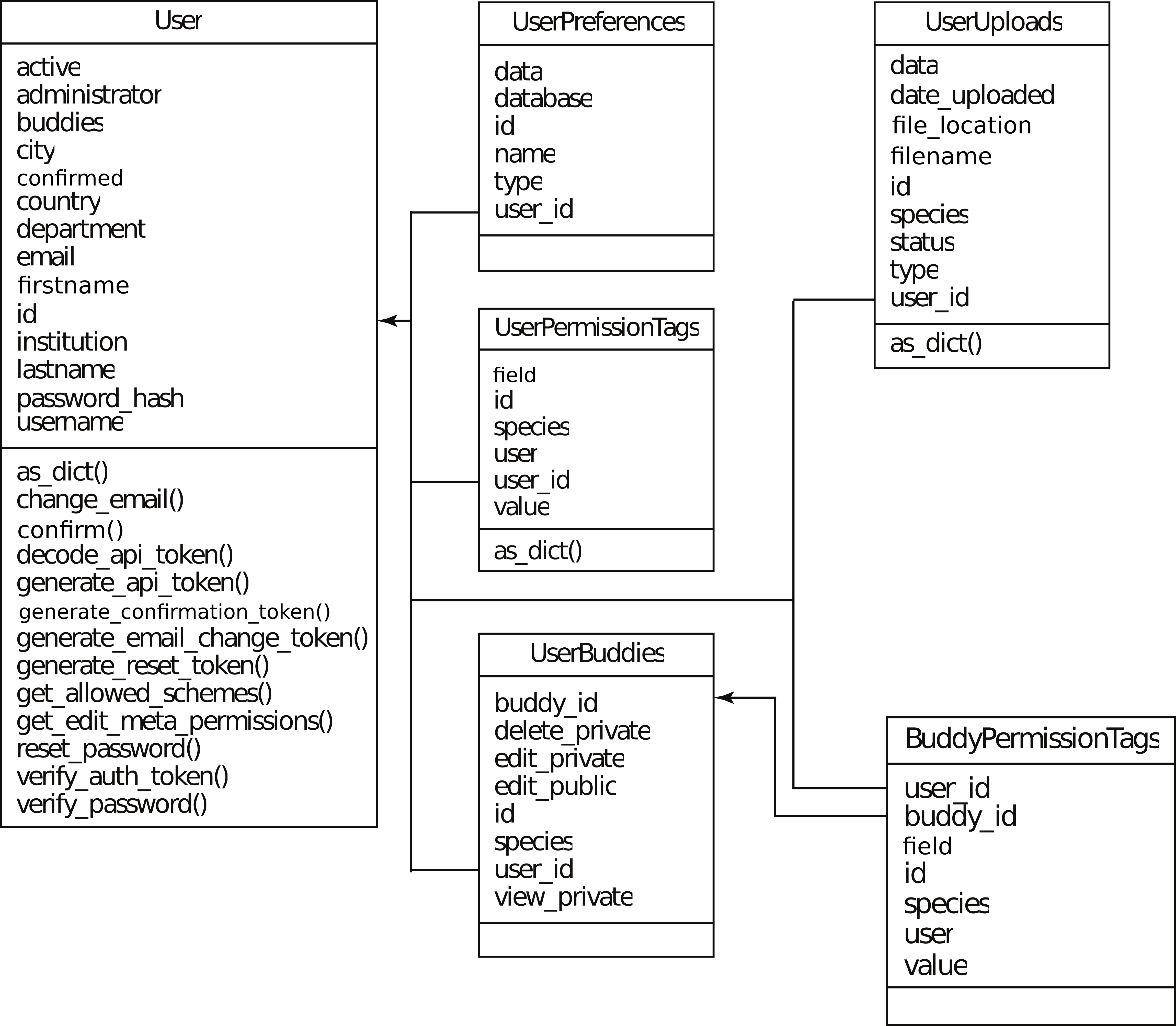
Figure 1: System database schematic
Species Database Structure
Each species in EnteroBase has its own database, with an identical table structure (Figure 2A). Fields in certain tables, namely Strains, are slightly different depending on the metadata required for different species. For instance, provision is made for ribotype in the Clostridioides database (Figure 2B).
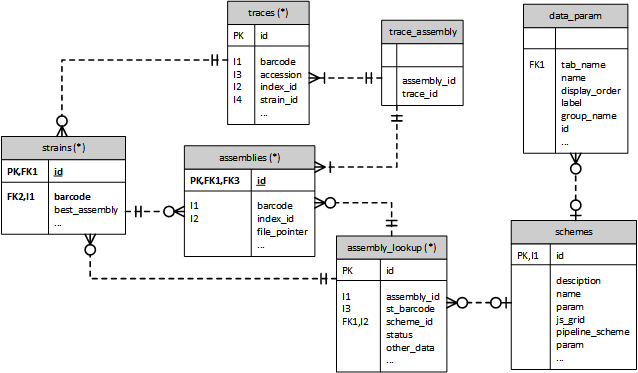
Figure 2: Species database schematic
The strain table is the master table for the metadata for each strain. The majority of the columns are common to all of the different species but there normally additional species specific columns in each species’ table. An entry in the strain table will normally be associated with a single trace entry providing information about the raw data used to create the assembly, as identified by the strain_id entry in the traces table. There may be more than one trace entry for a strain, for example if new read data is provided for the strain. When the trace is assembled an entry is created in the assemblies table and is then referenced by strains:best_assembly. The trace_assembly table identifies the assemblies associated with a trace, which does not appear to make sense as this allows for multiple traces to be associated with a single assembly whereas an assembly can only be associated with a single trace. There was originally a st_assembly table providing the link between strains and traces but this is no longer being updated so there is no direct connection from assemblies to strains.
When an assembly is succesfully created or uploaded, the pipeline jobs will then be run and an entry created in the assembly_lookup table for each assembly/pipeline job that has been run indicating whether the job has been run and whether it was succesful. This table provides the information used by the update_all_schemes process that is run every four hours that looks for outstanding pipeline jobs.
The schemes table is used to hold information associated with the data that is calculated for each strain. The most common entries are for the various schemes associated with the species, but the table hold information about other types of calculated data as well.
description The unique identifier associated with the scheme which should not be changed
name The label associated with the scheme that appears on the Experimental data dropdown
pipeline_scheme The identity of the pipeline job associated with the scheme. In the case of NServ/nomenclature jobs this is the name of the type of NServ job being run, the param:pipeline=’nomenclature’ json value indicates that a nomenclature pipeline job is associated with the scheme.
js_grid how the results are displayed
param A json entry that identifies various other miscellaneous information associated with the scheme, such as whether the results are displayed on the GUI.
The data_param table holds the information about each parameter that is generated associated with each scheme. This is linked to the scheme table by tabname==description. For results that are displayed it contains information about the way the parameter is displayed. For scheme data it holds information about each gene within the scheme. The relationship between these data and the data that are held within NServ is unclear. For information that is obtained from metaParser, the sra_field shows the identity of the data in the metaParser output
Versioning in EnteroBase
EnteroBase manages an internal log of all changes made to database records, particularly for data pertaining to strains, sequenced read traces, assemblies and genotyping. When these records are modified the current record is stamped with a version number, the time of the modification and the user who made the change. The previous state of the row is saved verbatim in an archive table. This provides a precise audit log of all changes in the database (Figure 2A).
Public API Structure
The API is implemented through the Flask web framework. A live demo of the API is available at https://enterobase.warwick.ac.uk/api/v2.0/swagger-ui . There are three generic classes that each specify how to handle requests for the following:
A single record (/api/v2.0/{database}/schemes/{barcode}),
Multiple records (/api/v2.0/{database}/schemes/),
And requests that have to be fetch internally from NServ (e.g. Sequence types)
Each API endpoint, e.g. ‘Schemes’ which is accessible through URLS like https://enterobase.warwick.ac.uk/api/v2.0/senterica/schemes, maps to a Resource class that define specific behaviours for processing different HTTP requests (GET, POST, PUT etc.) (Figure 4). These resource classes in turn have a Schema class that defines validation rules for API parameters, rules for mapping values to the correct database field and how to represent the final output (Figure 4).
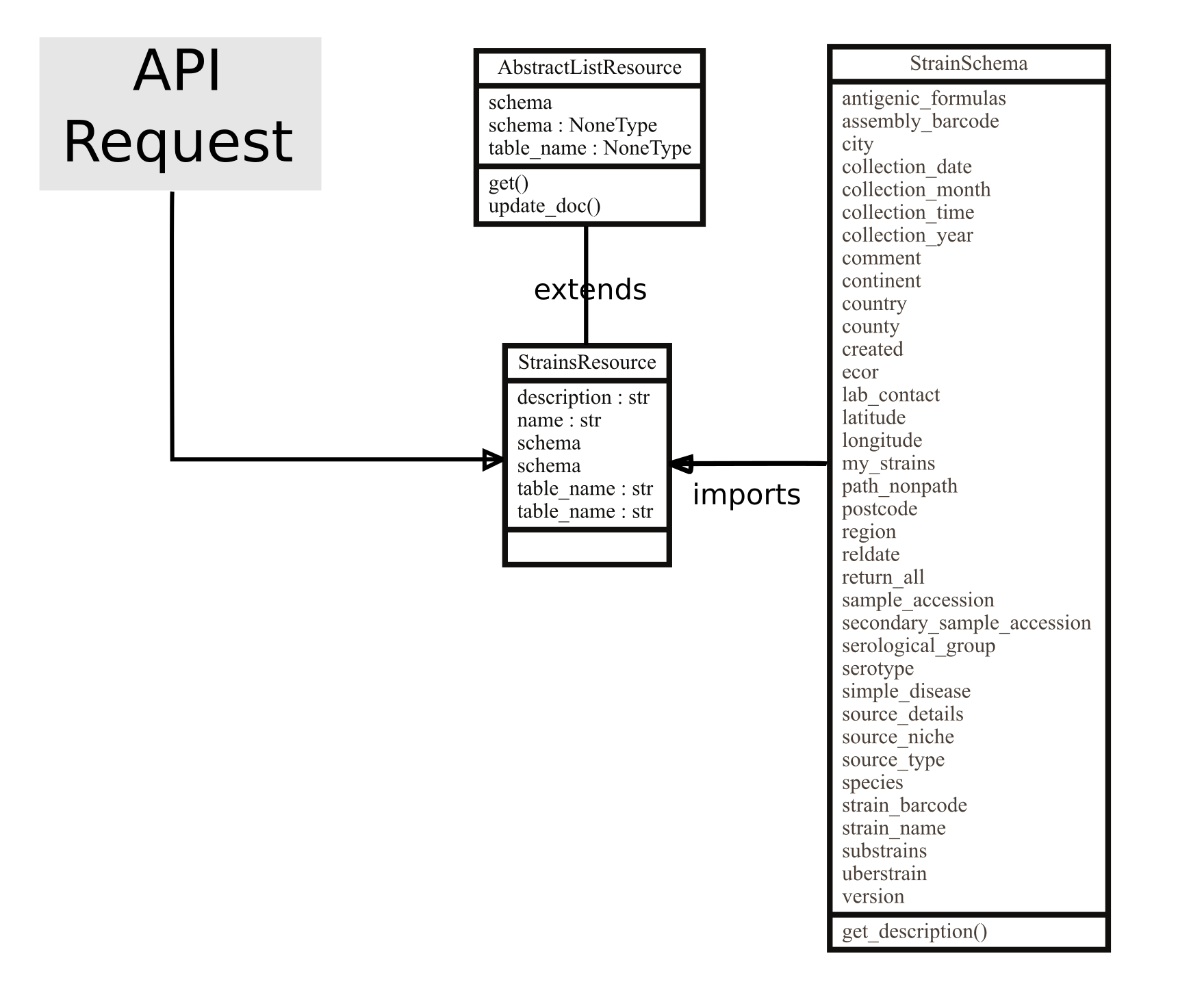
Figure 3: Basic interaction of API classes, using ‘Strains’ as an example.
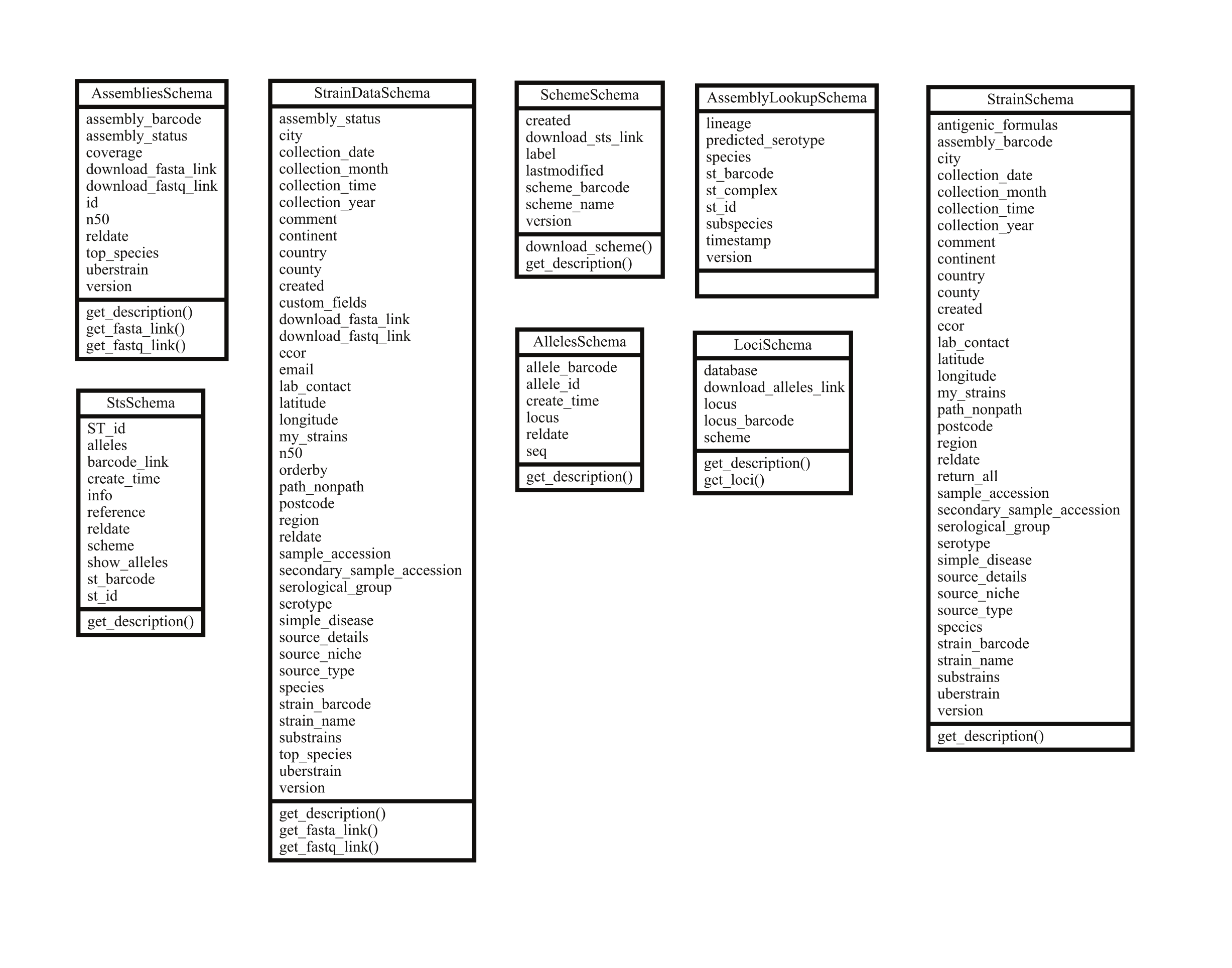
Figure 4: Attributes of schema for different API endpoints, these usually map to internal database fields.