EnteroBase & Bionumerics¶
We use Bionumerics 7.5+ for analysing data from EnteroBase.
By using the EnteroBase API you can do the same! This is a worked example showing you how to download genotyping data and strain metadata from EnteroBase into Bionumerics 7.5. This approach may work with 6, but I haven’t tried. You will need a valid API token, [click here for more details](Getting%20started%20with%20Enterobase%20API) For the sake of demonstration I will use a very small dataset of ~230 strains, which are UK Typhimurium strains from 2015 only.
For scripts, like these, that will help you to work with EnteroBase, [please click here] (https://bitbucket.org/enterobase/enterobase-scripts)
- Firstly, I will begin with a completely fresh BN 7.5 Database.
- We want to use BN’s in-built Python script editor. So go to Scripts > Python Script Editor…
Creating database fields¶
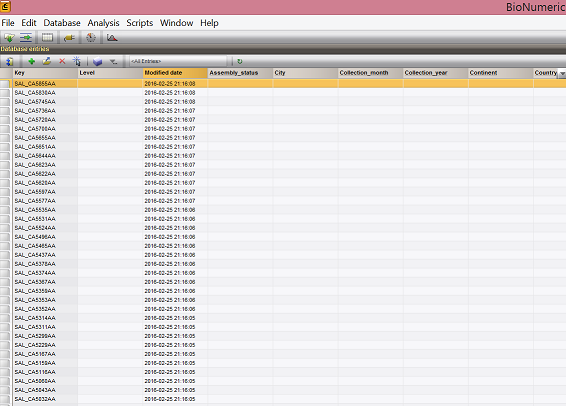
- The first thing we want to do is to populate the BN database with metadata fields. This is the template script with a few helper methods, and import_fields(), which creates the metadata fields.
Adding records¶
- **Second, we want to import some key values into the database. I prefer to use the strain BARCODE from EnteroBase for each record, it is unique across all EnteroBase databases, and excellent for searching a particular strain. The method that does this is import_entries() which is given the serotype, country and year we defined in __main__. **
def import_entries(serotype, country , year, max_num = 1000):
# Imports entry keys, given serotype, contact and year, into BN.
# This does not add any metadata.
offset = 0 # Cursor position for traversing data
limit = 200 # Page size in API requests
keys = [] # existing BN keys
for e in bns.Database.Db.Entries:
keys.append(str(e))
done = [] # BN does not add update keys until the end. So we need a
# running tally.
while True:
# Our search params
params = dict(Serovar=serotype, Country=country, Collection_year=year,
limit=limit, offset=offset, bionumerics='True')
# Retrive from strain data API
address = SERVER_ADDRESS + '/api/v1.0/%s/straindata?%s' %(DATABASE, urllib.urlencode(params))
print 'Connecting to %s' %address
response = urllib2.urlopen(create_request(address))
data = json.load(response)
# Read through data entries and add to BN.
for record in data['strains']:
item = record['strain_barcode']
if item not in keys and item not in done:
bns.Database.Db.Entries.Add(item)
done.append(item)
else:
print 'ERROR: %s has already been in this database' % (item)
offset += limit # Update cursor. Repeat.
# If the API does not give a link to a next page,
# this is the last page; so break.
if not data['paging'].has_key('next') or offset >= max_num:
break
- **The result is a database with just the keys. Metadata is loaded next. **
Adding Metadata¶
- **Metadata is loaded next with import_metadata(). I set it up to only update keys where the field Assembly_status is null, this field is always filled in EnteroBase, so I am implicitly picking rows that have no data at all. You can change the way the keys are chosen to anyway you want. **
def import_metadata(field_filter = 'Assembly_status', update_all = False):
# This method searches for rows missing data in a certain
# field and updates metadata.
offset = 0 # offset cursor for traversal
limit = 100 # page limit for API requests
keys = []
# Find all keys to be updated, where field is NULL.
for ent in bns.Database.Db.Entries:
if bns.Database.EntryField(ent,field_filter).Content == '' or update_all:
keys.append(str(ent))
fields = []
keys.sort()
# Fetch all BN fields to be updated.
for ent in bns.Database.Db.Fields:
fields.append(str(ent))
# Iterate through all keys to be updated
chunks =[keys[x:x+limit] for x in xrange(0, len(keys), limit)]
for chunk in chunks:
# Create list of strain keys to be downloaded
strain_list = ','.join(chunk)
# Request parameters
params = dict(strain_barcode=strain_list, bionumerics='True', limit=len(chunk))
address = SERVER_ADDRESS + '/api/v1.0/%s/straindata?%s' %(DATABASE, urllib.urlencode(params))
print 'Connecting to %s' %address[:50]
response = urllib2.urlopen(create_request(address))
data = json.load(response)
# From json data, update BN rows.
for record in data['strains']:
# Step through and updated specified fields.
for field in fields:
# Field should exist (obviously)
if record.has_key(field):
# Make sure API has returned a value for a field.
if record[field] != '' and record[field] != 'null' and record[field] != 'None' and record[field] != None:
# Do not update empty fields
if bns.Database.EntryField(record['strain_barcode'], field).Content != '' :
print "SKIP: EntryField '%s %s' has some data and will not be updated." % (record['strain_barcode'], field)
else :
bns.Database.EntryField(str(record['strain_barcode']), field).Content = str(record[field])
bns.Database.Db.Fields.Save() # commit our changes
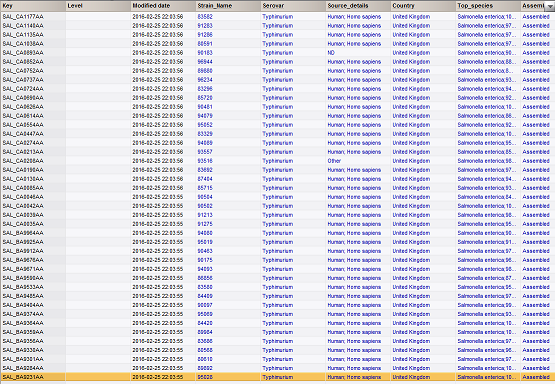
- **As an aside, you could choose keys based on rows you have selected in the main window with something like this **
- **Now we have something a bit more interesting in the database! **
Adding schemes¶
- The new step is to add the scheme definitions (experimental data) in BN
def create_experiments(schemes, maximum_num = 100000):
# Using list of schemes (from import fields)
# create corresponding experiments in BN and add loci.
keys = {}
# Valid schemes from import_fields method
for scheme in schemes:
# Fetch Loci information
address = SERVER_ADDRESS + '/api/v1.0/%s/%s/loci?limit=5000' %(DATABASE, scheme)
print 'Connecting to %s' %address
response = urllib2.urlopen(create_request(address))
data = json.load(response)
charset = []
for chars in data['loci']:
charset.append(chars['locus'])
# Get the scheme (experiment), or create if needed
try :
bns.Database.ExperimentType(scheme)
except :
bns.Database.Db.ExperimentTypes.Create(scheme,'CHR')
schemes[scheme] = bns.Characters.CharSetType(scheme)
# Create Locus (as a character field) if required
for cset in sorted(charset):
if schemes[scheme].FindChar(cset) == -1:
schemes[scheme].AddChar(cset, maximum_num)
# Add experimental data to keys for each scheme, if they have no ST info
for ent in bns.Database.Db.Entries:
ST = int(bns.Database.EntryField(ent,'%s_ST' %scheme).Content) if bns.Database.EntryField(ent,'%s_ST' %scheme).Content.isdigit() else 100000
# Records with invalid or undefined STs will be given a -1 value.
# These need to be ignored.
if ST < 1:
bns.Database.Experiment(ent, scheme_name).Delete()
elif bns.Database.EntryField(ent,'%s_ST' %scheme).Content == '':
keys[str(ent)] = True
return sorted(keys.keys())
- ** Schemes are now added, with all the loci as well.**
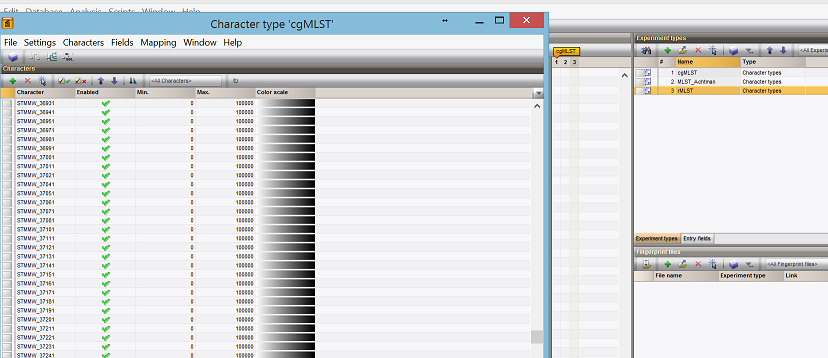
Loading genotype data¶
- ** Now the fun part. Loading the actual genotyping data in for all schemes, including cgMLST**
def get_experiment_data(keys):
limit = 100
# Iterate through all keys (in chunks) to be updated
chunks =[keys[x:x+limit] for x in xrange(0, len(keys), limit)]
cset = bns.Characters.CharSet()
for chunk in chunks:
# Create list of strain keys to be downloaded
strain_list = ','.join(chunk)
# Request parameters
params = dict(strain_barcode=strain_list, bionumerics='True', limit=len(chunk))
address = SERVER_ADDRESS + '/api/v1.0/%s/straindata?%s' %(DATABASE, urllib.urlencode(params))
# Fetch records
response = urllib2.urlopen(create_request(address))
data = json.load(response)
for record in data['strains']:
# Retrieve ST information for records.
# Iterate through sts, which stores ST info for each scheme
for st in record['sts']:
if st.has_key('st_barcode_link') and st['st_barcode'].endswith('_ST'):
ST = None
# Search for an ST for the records
scheme_name = st['scheme_name']
try:
lookup_data = json.load(urllib2.urlopen(create_request(st['st_barcode_link'])))
# Request may fail and return an error message (as a string), ignore.
if isinstance(lookup_data['results'][0], unicode):
print 'ERROR: parsing lookup data'
elif lookup_data['results'][0].has_key('ST_id'):
ST = str(lookup_data['results'][0]['ST_id'])
except Exception as e:
print 'ERROR: parsing lookup data %s ' %e
if ST:
# Update ST value if exists
bns.Database.EntryField(str(record['strain_barcode']), scheme_name + '_ST').Content = ST
# Add character profile (allele numbers) if ST > 0 (valid)
if int(ST) > 0 :
# Create Experiment record if required
if not cset.Load(record['strain_barcode'], scheme_name) :
cset.Create(scheme_name, '', record['strain_barcode'])
# Add Allele profile
for allele in lookup_data['results'][0]['alleles']:
id = cset.FindName(allele['locus'])
if id >= 0 :
cset.SetVal(id, float(allele['allele_id']))
cset.Save() # Commit changes
else:
# ST is not valid, delete data if any.
bns.Database.Experiment(record['strain_barcode'], scheme_name).Delete()
# Fetch associated data (eBG, lineage, Predicted serovar). Lookup ST.
eBG_address = SERVER_ADDRESS + '/api/v1.0/%s/%s/sts?ST_id=%s' %(DATABASE, scheme_name, ST)
try:
eBG_data = json.load(urllib2.urlopen(create_request(eBG_address)))
# Request may fail and return error, valid data is always a dict
if isinstance(eBG_data,dict):
eBG_info = None
if len(eBG_data.get('STs')) > 0:
eBG_info = eBG_data.get('STs')[0].get('info')
# Found valid associated data, adding...
if eBG_info:
eBG = eBG_info.get('st_complex','')
lineage = eBG_info.get('lineage','')
serovars = eBG_info.get('predict',{}).get('serotype',[])
if len(serovars) > 0:
prediction = serovars[0][0]
bns.Database.EntryField(str(record['strain_barcode']), 'Predicted_serovar').Content = prediction
bns.Database.EntryField(str(record['strain_barcode']), scheme_name + '_eBG').Content = eBG
bns.Database.EntryField(str(record['strain_barcode']), 'Lineage').Content = lineage
else:
print 'ERROR: %s has no eBG info' %record['strain_barcode']
except:
print 'Error on eBG lookup ST_id: %s' % ST
else:
# No ST could be found in Lookup.
print 'ERROR: %s has no ST profile data' %record['strain_barcode']
bns.Database.Experiment(record['strain_barcode'], st['scheme_name']).Delete()
bns.Database.Db.Fields.Save() # Commit changes
- ** Grab a coffee, sit back and watch the little green blips light up as data is loaded in. **
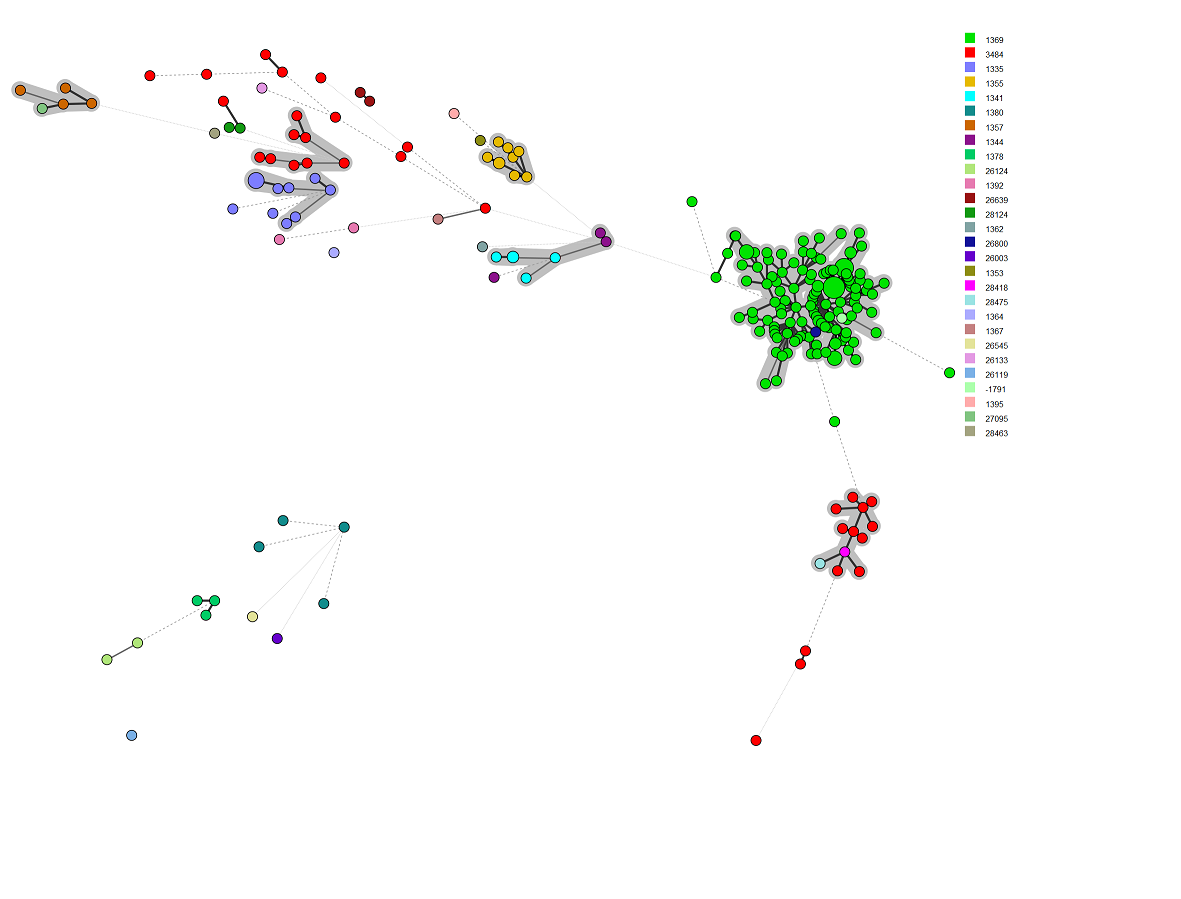
The final MST¶
** Congratulations! You’ve got strain metadata, cgMLST, rMLSt, and 7 Gene MLST data loaded into BN. Here is the MST generated from these data. I have colour coded it with rMLST ST. I set the partitions [grey] at 100 allele differences. **
The demo script¶
- ** Here is the entire demo script: **
import os
import sys
import site
import urllib
import urllib2
import json
import base64
import bns
# Created by Nabil-Fareed Alikhan 2015 <enterobase@warwick.ac.uk>
# You have a choice of Databases; 'senterica', 'ecoli', 'yersinia', 'mcatarrhalis'
# for Salmonella, Escherichia, Yersinia and Moraxella database respectively.
DATABASE = 'senterica'
SERVER_ADDRESS = 'https://enterobase.warwick.ac.uk/'
# This should be the Enterobase API Token from the website.
TOKEN = ''
def create_request(request_str):
''' Helper method that formats a URL request for the API by
wrapping it with your token
'''
request = urllib2.Request(request_str)
base64string = base64.encodestring('%s:%s' % (TOKEN,'')).replace('\n', '')
request.add_header("Authorization", "Basic %s" % base64string)
return request
def import_fields():
''' Creates fields and experimental data types,
using one strain record as a template.
Returns list of scheme names, required later.
'''
# URL for EnteroBase strain records. Fetch 1 only.
address = SERVER_ADDRESS + '/api/v1.0/%s/straindata?bionumerics=True&limit=1&Assembly_status=Assembled' %DATABASE
print 'Connecting to %s' %address
response = urllib2.urlopen(create_request(address))
# Strain data is returned as json.
data = json.load(response)
record = data["strains"][0]
existing_fields = []
# Add metadata fields from EnteroBase to BN.
for e in bns.Database.Db.Fields:
existing_fields.append(str(e))
for key in sorted(record.keys()):
# Clean up any weird characters.
new_key = key.replace('.','_')
new_key = new_key.replace(' ','_')
# There are helper fields (like url links or nested data in EnteroBase output,
# ignore these fields i.e. 'link' and 'sts'
if not new_key.endswith('link') and not new_key.startswith('sts') and new_key not in existing_fields:
bns.Database.Db.Fields.Add(new_key)
# Add schemes to BN.
schemes = {}
for st in sorted(record['sts']):
scheme_name = st['scheme_name']
# We only want 7 Gene MLST, cgMLST or rMLST data right now.
if scheme_name.startswith('MLST') or scheme_name in ['cgMLST','rMLST']:
schemes[scheme_name] = st['scheme_id']
# Add fields for ST and eBG
st_field = '%s_ST' %scheme_name.replace('.','_').replace(' ','_')
ebg_field = '%s_eBG' %scheme_name.replace('.','_').replace(' ','_')
if st_field not in existing_fields:
bns.Database.Db.Fields.Add(st_field)
if ebg_field not in existing_fields:
bns.Database.Db.Fields.Add(ebg_field)
# Add a field for Predicted serovar.
if 'Predicted_serovar' not in existing_fields:
bns.Database.Db.Fields.Add('Predicted_serovar')
# Commit our changes
bns.Database.Db.Fields.Save()
return schemes
def import_entries(serotype, country , year, max_num = 1000):
# Imports entry keys, given serotype, contact and year, into BN.
# This does not add any metadata.
offset = 0 # Cursor position for traversing data
limit = 200 # Page size in API requests
keys = [] # existing BN keys
for e in bns.Database.Db.Entries:
keys.append(str(e))
done = [] # BN does not add update keys until the end. So we need a
# running tally.
while True:
# Our search params
params = dict(Serovar=serotype, Country=country, Collection_year=year,
limit=limit, offset=offset, bionumerics='True')
# Retrive from strain data API
address = SERVER_ADDRESS + '/api/v1.0/%s/straindata?%s' %(DATABASE, urllib.urlencode(params))
print 'Connecting to %s' %address
response = urllib2.urlopen(create_request(address))
data = json.load(response)
# Read through data entries and add to BN.
for record in data['strains']:
item = record['strain_barcode']
if item not in keys and item not in done:
bns.Database.Db.Entries.Add(item)
done.append(item)
else:
print 'ERROR: %s has already been in this database' % (item)
offset += limit # Update cursor. Repeat.
# If the API does not give a link to a next page,
# this is the last page; so break.
if not data['paging'].has_key('next') or offset >= max_num:
break
def import_metadata(field_filter = 'Assembly_status', update_all = False):
# This method searches for rows missing data in a certain
# field and updates metadata.
offset = 0 # offset cursor for traversal
limit = 100 # page limit for API requests
keys = []
# Find all keys to be updated, where field is NULL.
for ent in bns.Database.Db.Entries:
if bns.Database.EntryField(ent,field_filter).Content == '' or update_all:
keys.append(str(ent))
fields = []
keys.sort()
# Fetch all BN fields to be updated.
for ent in bns.Database.Db.Fields:
fields.append(str(ent))
# Iterate through all keys to be updated
chunks =[keys[x:x+limit] for x in xrange(0, len(keys), limit)]
for chunk in chunks:
# Create list of strain keys to be downloaded
strain_list = ','.join(chunk)
# Request parameters
params = dict(strain_barcode=strain_list, bionumerics='True', limit=len(chunk))
address = SERVER_ADDRESS + '/api/v1.0/%s/straindata?%s' %(DATABASE, urllib.urlencode(params))
print 'Connecting to %s' %address[:50]
response = urllib2.urlopen(create_request(address))
data = json.load(response)
# From json data, update BN rows.
for record in data['strains']:
# Step through and updated specified fields.
for field in fields:
# Field should exist (obviously)
if record.has_key(field):
# Make sure API has returned a value for a field.
if record[field] != '' and record[field] != 'null' and record[field] != 'None' and record[field] != None:
# Do not update empty fields
if bns.Database.EntryField(record['strain_barcode'], field).Content != '' :
print "SKIP: EntryField '%s %s' has some data and will not be updated." % (record['strain_barcode'], field)
else :
bns.Database.EntryField(str(record['strain_barcode']), field).Content = str(record[field])
bns.Database.Db.Fields.Save() # commit our changes
def create_experiments(schemes, maximum_num = 100000):
# Using list of schemes (from import fields)
# create corresponding experiments in BN and add loci.
keys = {}
# Valid schemes from import_fields method
for scheme in schemes:
# Fetch Loci information
address = SERVER_ADDRESS + '/api/v1.0/%s/%s/loci?limit=5000' %(DATABASE, scheme)
print 'Connecting to %s' %address
response = urllib2.urlopen(create_request(address))
data = json.load(response)
charset = []
for chars in data['loci']:
charset.append(chars['locus'])
# Get the scheme (experiment), or create if needed
try :
bns.Database.ExperimentType(scheme)
except :
bns.Database.Db.ExperimentTypes.Create(scheme,'CHR')
schemes[scheme] = bns.Characters.CharSetType(scheme)
# Create Locus (as a character field) if required
for cset in sorted(charset):
if schemes[scheme].FindChar(cset) == -1:
schemes[scheme].AddChar(cset, maximum_num)
# Add experimental data to keys for each scheme, if they have no ST info
for ent in bns.Database.Db.Entries:
ST = int(bns.Database.EntryField(ent,'%s_ST' %scheme).Content) if bns.Database.EntryField(ent,'%s_ST' %scheme).Content.isdigit() else 100000
# Records with invalid or undefined STs will be given a -1 value.
# These need to be ignored.
if ST < 1:
bns.Database.Experiment(ent, scheme_name).Delete()
elif bns.Database.EntryField(ent,'%s_ST' %scheme).Content == '':
keys[str(ent)] = True
return sorted(keys.keys())
def get_experiment_data(keys):
limit = 100
# Iterate through all keys (in chunks) to be updated
chunks =[keys[x:x+limit] for x in xrange(0, len(keys), limit)]
cset = bns.Characters.CharSet()
for chunk in chunks:
# Create list of strain keys to be downloaded
strain_list = ','.join(chunk)
# Request parameters
params = dict(strain_barcode=strain_list, bionumerics='True', limit=len(chunk))
address = SERVER_ADDRESS + '/api/v1.0/%s/straindata?%s' %(DATABASE, urllib.urlencode(params))
# Fetch records
response = urllib2.urlopen(create_request(address))
data = json.load(response)
for record in data['strains']:
# Retrieve ST information for records.
# Iterate through sts, which stores ST info for each scheme
for st in record['sts']:
if st.has_key('st_barcode_link') and st['st_barcode'].endswith('_ST'):
ST = None
# Search for an ST for the records
scheme_name = st['scheme_name']
try:
lookup_data = json.load(urllib2.urlopen(create_request(st['st_barcode_link'])))
# Request may fail and return an error message (as a string), ignore.
if isinstance(lookup_data['results'][0], unicode):
print 'ERROR: parsing lookup data'
elif lookup_data['results'][0].has_key('ST_id'):
ST = str(lookup_data['results'][0]['ST_id'])
except Exception as e:
print 'ERROR: parsing lookup data %s ' %e
if ST:
# Update ST value if exists
bns.Database.EntryField(str(record['strain_barcode']), scheme_name + '_ST').Content = ST
# Add character profile (allele numbers) if ST > 0 (valid)
if int(ST) > 0 :
# Create Experiment record if required
if not cset.Load(record['strain_barcode'], scheme_name) :
cset.Create(scheme_name, '', record['strain_barcode'])
# Add Allele profile
for allele in lookup_data['results'][0]['alleles']:
id = cset.FindName(allele['locus'])
if id >= 0 :
cset.SetVal(id, float(allele['allele_id']))
cset.Save() # Commit changes
else:
# ST is not valid, delete data if any.
bns.Database.Experiment(record['strain_barcode'], scheme_name).Delete()
# Fetch associated data (eBG, lineage, Predicted serovar). Lookup ST.
eBG_address = SERVER_ADDRESS + '/api/v1.0/%s/%s/sts?ST_id=%s' %(DATABASE, scheme_name, ST)
try:
eBG_data = json.load(urllib2.urlopen(create_request(eBG_address)))
# Request may fail and return error, valid data is always a dict
if isinstance(eBG_data,dict):
eBG_info = None
if len(eBG_data.get('STs')) > 0:
eBG_info = eBG_data.get('STs')[0].get('info')
# Found valid associated data, adding...
if eBG_info:
eBG = eBG_info.get('st_complex','')
lineage = eBG_info.get('lineage','')
serovars = eBG_info.get('predict',{}).get('serotype',[])
if len(serovars) > 0:
prediction = serovars[0][0]
bns.Database.EntryField(str(record['strain_barcode']), 'Predicted_serovar').Content = prediction
bns.Database.EntryField(str(record['strain_barcode']), scheme_name + '_eBG').Content = eBG
bns.Database.EntryField(str(record['strain_barcode']), 'Lineage').Content = lineage
else:
print 'ERROR: %s has no eBG info' %record['strain_barcode']
except:
print 'Error on eBG lookup ST_id: %s' % ST
else:
# No ST could be found in Lookup.
print 'ERROR: %s has no ST profile data' %record['strain_barcode']
bns.Database.Experiment(record['strain_barcode'], st['scheme_name']).Delete()
bns.Database.Db.Fields.Save() # Commit changes
if __name__ == '__main__':
schemes = import_fields()
serotype = 'Typhimurium'
country = 'United Kingdom'
year = '2015'
import_entries(serotype, country, year)
import_metadata()
keys_missing_exp = create_experiments(schemes)
get_experiment_data(keys_missing_exp)
Appendix 1: Sample strain record.¶
This a sample of what a strain record looks like coming from EnteroBase, if you want to write your own searches. Follow the ST barcode link for all the ST information.
{
"secondary_sample_accession": "SRS1045120",
"City": null,
"antigenic_formulas": null,
"metadata_version": 1,
"assembly_barcode": "SAL_EA1761AA_AS",
"Country": "United States",
"Region": null,
"Collection_month": null,
"sts": [
{
"st_barcode_link": "https://enterobase.warwick.ac.uk/api/v1.0/lookup?barcode=SAL_RA4950AA_ST",
"scheme_id": 6,
"scheme_name": "cgMLST",
"st_version": "3.1",
"st_barcode": "SAL_RA4950AA_ST"
},
{
"st_barcode_link": "https://enterobase.warwick.ac.uk/api/v1.0/lookup?barcode=SAL_AA0015AA_ST",
"scheme_id": 1,
"scheme_name": "MLST_Achtman",
"st_version": "3.0",
"st_barcode": "SAL_AA0015AA_ST"
},
{
"st_barcode_link": "https://enterobase.warwick.ac.uk/api/v1.0/lookup?barcode=SAL_AA4248AA_ST",
"scheme_id": 3,
"scheme_name": "rMLST",
"st_version": "3.0",
"st_barcode": "SAL_AA4248AA_ST"
}
],
"Assembly_status": "Assembled",
"Source_details": null,
"last_modified": "2015-09-03 14:49:49.651160",
"secondary_study_accession": "SRP040281",
"Top_species": "Salmonella enterica;100.0%",
"strain_barcode": "SAL_AA7051AA",
"Email": "pfge@cdc.gov",
"Strain_Name": "PNUSAS000536",
"Continent": "North America",
"Source_niche": null,
"release_date": "2015-08-25 00:00:00",
"sample_accession": "SAMN03863381",
"County": null,
"Source_type": null,
"Serovar": null,
"Lab_contact": "EDLB-CDC",
"assembly_version": 1,
"Collection_year": null,
"study_accession": "PRJNA230403"
}