EnteroBase QAssembly¶
QAssembly (high Q**uality **Assembly) is an assembly pipeline that aims to generate currently best assemblies within a reasonable amount of time.
The whole pipeline (current version: 3.61) contains several components:
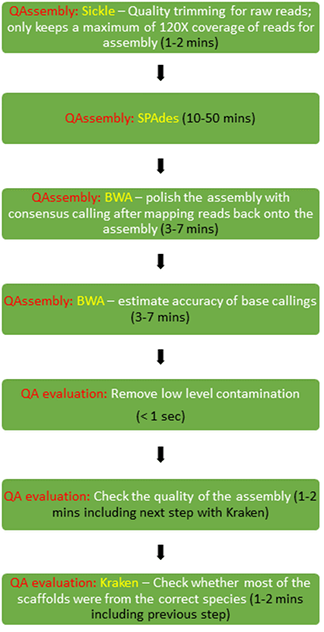
Quality Trimming (Sickle) & Barcode trimming¶
As recommended by Del Fabbro *et al.*, QAssembly uses Sickle 1.33 with the argument -q 10 on FASTQ files to trim the ends of short reads of base calls with quality scores = 10.
Some very old QAIIx reads, or reads generated for special experiments contain barcodes at their 5’ ends in order to mark multiple biological samples within a common sequencing pool. Such barcodes result in the presence of identical runs of 5’ nucleotides in >=50% of reads, which were identified 2 bp at a time and stripped by a nested Python function called read_process(). This function also removes reads from SRAs with too many reads, limiting their maximum size to to > 200X of the genome size. This approach was recommended by Andrew Millard’s blog in order to reduce un-necessary processing time and memory because the quality of assemblies does not improve once read depths exceed 30X genome coverage.
SPAdes Assembly¶
Assembly is performed with SPAdes 3.9.0 on 7 threads, without pre-correction with BayesHammer. BayesHammer did not provide significant improvements in benchmark comparisons, and doubled the time needed for assemblies. We also did not use post-polishing within SPAdes because it is inferior to the post-polishing steps in QAssembly (below).
BWA Remapping and base correction¶
Raw reads are mapped back onto assembly scaffolds in order to improve the accuracy of consensus base calls.
- BWA 0.7.12-r1039 is used to align reads back onto the assemblies.
- Because BWA was developed for alignments between genetically distinct sequences, it does not necessarily align the entirety of all reads against the consensus assembly, especially if it contains local mis-assemblies by SPAdes due to weak connections. These were identified by extending the BWA alignments over the entire length of the reads with the help of Python script (sam_filter.py) in order to mark such mis-assemblies for the next step.
- Consensus base calling, including format transformation, are performed with SAMtools and BCFtools (both in version 1.2). The prior assemblies are then corrected (polished) by incorporating differing, most probable consensus bases or indels reported by bcftools call.
BWA base quality and save in FASTQ format¶
Base-specific quality scores are called a second time, in order to assess the uncertainties of the consensus callings in the assemblies. BWA and SAMtools and BCFtools are used to generate and analyse the remappings. The qualities of consensus bases were given by comparing the supports to the consensus callings against all other alternatives.
Conversion of QAFastq format into FASTA format¶
The :doc`QAtoFasta <backend-pipeline-qatofasta` pipeline processes the assemblies generated by QAssembly in QAFastq format in order to convert the assemblies to FASTA format.
Quality Control¶
The QA evaluation pipeline, run as part of the QAssembly pipeline is described in Quality Assessment evaluation. Assemblies are evaluated according to several criteria described on that page (including a check on species assignment of contigs with Kraken). Any assembly that is failed in this quality control will not be used to call MLST or for other downstream analyses.
The QAEvaluation pipeline, also processes the assemblies in order to mask the sequence. Masking the sequence consists of replacing bases in the sequence whose base qualities are below a cutoff (10) with the IUPAC code for any base (i.e. “N”). Masked sequence for the assemblies in FASTA format is made available for users to download (and may be available even if the assemblies failed QC providing that the earlier assembly stage did not fail itself).